- 广济众生 致力人类健康
2018年5月8日是第25个世界地贫日,
“防治地贫 健康脱贫”是今年随处可见的主题。
对于“地中海贫血”,
你只听说过它的“可怕”,
却没有真正地走入它的“世界”。
地中海贫血的历史
1925年,意大利医生Cooley首次报道5例具有严重贫血、脾脏肿大、异常的骨骼病变以及外周血出现大量幼稚红细胞的小儿病例, 由于所有病人均来自地中海之滨的意大利和希腊,Cooley将其称之为“地中海贫血”
地中海贫血的概述
原名地中海贫血又称海洋性贫血,是一组遗传性溶血性贫血疾病。由于遗传的基因缺陷致使血红蛋白中一种或一种以上珠蛋白链合成缺如或不足所导致的贫血或病理状态。缘于基因缺陷的复杂性与多样性,使缺乏的珠蛋白链类型、数量及临床症状变异性较大。根据所缺乏的珠蛋白链种类及缺乏程度予以命名和分类。
地中海贫血基因的来源
假说:是人类对抗疟疾而发生基因突变,自然选择的结果
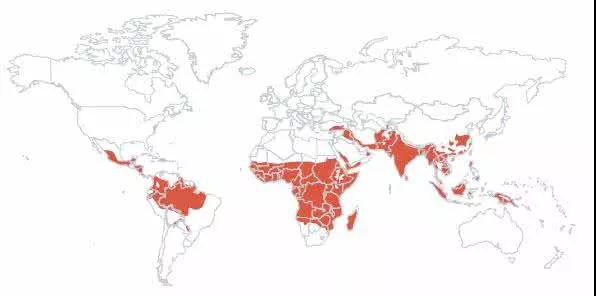
疟疾分布图
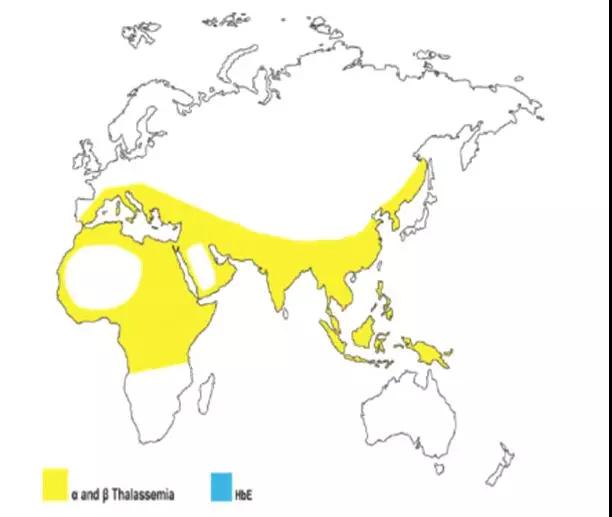
地贫分布图
地中海贫血病因
珠蛋白链的分子结构及合成是由基因决定的。γ、δ、ε和β珠蛋白基因组成“β基因族”,ζ和α珠蛋白组成“α基因族”。正常人自父母双方各继承2个α珠蛋白基因(αα/αα)合成足够的α珠蛋白链;自父母双方各继承1个β珠蛋白基因合成足够的β珠蛋白链。由于珠蛋白基因的缺失或点突变,肽链合成障碍导致发病。地中海贫血分为α型、β型、δβ型和δ型4种,其中以β和α地中海贫血较为常见。
1.β珠蛋白生成障碍性贫血(β地中海贫血)
β珠蛋白生成障碍性贫血(简称β地贫)的发生的分子病理相当复杂,已知有100种以上的β基因突变,主要是由于基因的点突变,少数为基因缺失。
2.α珠蛋白生成障碍性贫血(α地中海贫血)
大多数α珠蛋白生成障碍性贫血(地中海贫血)(简称α地贫)是由于α珠蛋白基因的缺失所致,少数由基因点突变造成。白基因的缺失所致,少数由基因点突变造成。
地中海贫血临床表现
根据病情轻重的不同,分为以下3型。
1. 重型
出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型的表现是臀状头,长骨可骨折。骨骼改变是骨髓造血功能亢进、骨髓腔变宽、皮质变薄所致。少数患者在肋骨及脊椎之间发生胸腔肿块,亦可见胆石症、下肢溃疡。
2. 中间型
轻度至中度贫血,患者大多可存活至成年。
3. 轻型
轻度贫血或无症状,一般在调查家族史时发现。
重在预防!!!
地贫是严重致死、致残性遗传性血液病
1. 贫血:组织供氧不足,功能衰退,危及生命;促进机体造血
2. 骨骼畸形:骨髓过度造血;骨板变薄,骨质疏松;骨骼畸形,特殊面容
3. 多脏器损伤:组织缺氧损伤;消化系统过度吸收铁;铁过度沉淀引起这些器官功能衰竭而死亡
地贫患儿给家庭和社会带来巨大负担!
地贫患儿的出生严重影响人口素质,并威胁人类生命安全。
经济损失巨大(广东省妇幼保健院研究表明,一个地贫儿平均导致300万损失)
一级预防:婚前及孕前检查(遗传咨询)
二级预防:产前检查与诊断(是在孕期通过早发现、早诊断和早采取干预措施,以预防重度地贫患儿的出生)
三级预防:阳性新生儿检查(胎儿宫内治疗、新生儿治疗)
一般来说,如果两名属同一类型的地中海贫血患者结合,便有机会生下重型贫血患者。要想有效预防本病,需抽血进行肽链检测和基因分析,若证实本身和配偶同属β型极轻型或轻型地贫患者,子女将有四分之一的机会完全正常、二分之一的机会成为轻型贫血患者,四分之一的机会成为中型或重型贫血患者。鉴于本病缺少根治的方法,临床中、重型预后不良,故在婚配方面医生应向有阳性家族史或患者提出医学建议,进行婚前检查和胎儿产前基因诊断,避免下一代患儿的发生。
